Authors and Disclosures
Posted: 10/28/2011; Pract Neurol. 2011;11(5):272-282
Abstract and Introduction
Abstract
Stiff person syndrome (SPS) is a rare disorder, characterised by fluctuating rigidity and stiffness of the axial and proximal lower limb muscles, with superimposed painful spasms and continuous motor unit activity on electromyography. Although rare in general neurology practice, once observed it is unforgettable. The general neurologist may see only one or two cases during his or her career and as such it remains underdiagnosed. Left untreated, SPS symptoms can progress to cause significant disability. Patients have a poor quality of life and an excess rate of comorbidity and mortality. The severity of symptoms and lack of public awareness of the condition create anxiety and uncertainty for people with the disease. This review aims to raise awareness of SPS and to improve the likelihood of its earlier diagnosis and treatment.
Introduction
Stiff person syndrome (SPS) is rare, although its true frequency has not been fully ascertained. The British Neurological Surveillance Unit identified 119 cases among the UK population over 5 years (2000–2005), implying a prevalence of 1–2 cases per million.
The natural history of SPS has yet to be completely described. The symptoms range from mild to severe and can progress, resulting in significant disability. Perhaps 65% of patients cannot independently perform normal activities of daily living due to rigidity and stiffness, phobias, unpredictable spasms and frequent falls.[1]
Almost all SPS has an autoimmune basis; most have the glutamic acid decarboxylase (GAD) autoantibody but there are also paraneoplastic varieties. Main line therapies include γ-aminobutyric acid (GABA)-ergic and other antispasmodic agents to alleviate symptoms, but immunomodulatory agents can also attenuate the aberrant immune process.
Background
Moersch and Woltman described 'stiff man syndrome' in 1956 in 14 patients with tightness of the back, abdominal and thigh muscles.[2] Their observations spanned a 32-year period and they detailed progressive fluctuating rigidity and painful spasms, leading to a characteristic 'wooden man' appearance, with postural instability and falls. Diabetes mellitus (DM) accompanied a handful of cases.
Eleven years later, Gordon et al set out clinical criteria to address the need for improved diagnostic certainty.[3] These were modified by Lorish et al in 1989[4] and by Dalakas in 2009.[1] These latest criteria are currently in use. Moersch and Woltman described the electromyographic findings of motor unit activity resembling 'that which accompanies contraction of voluntary muscle'.[2] Gordon and colleagues (1967) used electromyography and muscle relaxants, describing 'persistent tonic contraction reflected in constant firing, even at rest'.[3]
In 1988, Solimena et al described the major breakthrough in the pathogenesis of SPS in a patient presenting with diabetic ketoacidosis. Through elegant experiments, they identified an autoimmune link between SPS and DM. They proposed that autoantibodies against GAD, an enzyme found in both the central nervous system and in pancreatic islets of Langerhans, were important to both diseases.[5] The GAD enzyme was already known to synthesise the inhibitory neurotransmitter, GABA; their findings provided a plausible disease mechanism. Of interest, Howard had reported previously that diazepam significantly reduced muscle stiffness and rigidity in SPS, suggesting involvement of GABA-related inhibitory pathways (figure 1).[6]
Eleven years later, Gordon et al set out clinical criteria to address the need for improved diagnostic certainty.[3] These were modified by Lorish et al in 1989[4] and by Dalakas in 2009.[1] These latest criteria are currently in use. Moersch and Woltman described the electromyographic findings of motor unit activity resembling 'that which accompanies contraction of voluntary muscle'.[2] Gordon and colleagues (1967) used electromyography and muscle relaxants, describing 'persistent tonic contraction reflected in constant firing, even at rest'.[3]
In 1988, Solimena et al described the major breakthrough in the pathogenesis of SPS in a patient presenting with diabetic ketoacidosis. Through elegant experiments, they identified an autoimmune link between SPS and DM. They proposed that autoantibodies against GAD, an enzyme found in both the central nervous system and in pancreatic islets of Langerhans, were important to both diseases.[5] The GAD enzyme was already known to synthesise the inhibitory neurotransmitter, GABA; their findings provided a plausible disease mechanism. Of interest, Howard had reported previously that diazepam significantly reduced muscle stiffness and rigidity in SPS, suggesting involvement of GABA-related inhibitory pathways (figure 1).[6]
 Figure 1. The timeline of significant dates in the history of SPS.[2 5–12] GABARAP, γ-aminobutyric acid (A) receptor-associated protein; GAD, glutamic acid decarboxylase; SPS, stiff person syndrome
Figure 1. The timeline of significant dates in the history of SPS.[2 5–12] GABARAP, γ-aminobutyric acid (A) receptor-associated protein; GAD, glutamic acid decarboxylase; SPS, stiff person syndromePathophysiology
The exact pathogenic role of autoantibodies in SPS remains unclear. All SPS autoantigens identified to date are synaptic proteins involved in inhibitory synaptic transmission. The presynaptic autoantigens are GAD and amphiphysin, and the postsynaptic autoantigens are GABA (A) receptor-associated protein and gephyrin (figure 2).[5 10–12]
The SPS autoantibodies probably mediate their effect through pharmacological blockade of their target autoantigens rather than by causing structural changes in GABA-ergic neurons. This is supported by the lack of abnormal neurological signs other than increased muscle tone, and the improvement of symptoms with immunotherapy.[13] The fact that most cases have normal postmortem findings supports this theory. Some cases show non-specific neuronal and interneuronal loss in the spinal cord.[14]
Interestingly, a report of stiff horse syndrome described a stiff gait and painful muscles to palpation. The clinical examination, electromyographic and serological investigations were strikingly similar to SPS.[15]
GAD Autoantibodies
About 60–80% of SPS cases have autoantibodies against GAD, a rate-limiting cytoplasmic enzyme responsible for synthesising the inhibitory neurotransmitter GABA in the brain and spinal cord.[1 16] GAD is synthesised mainly in presynaptic GABA-ergic neurons in both the central nervous system and in the islet of Langerhans β-cells of the pancreas (figure 2).[16] The mechanism by which autoantibodies recognise intracellular GAD is unclear.
There are two GAD isoforms—GAD65 and GAD67—but it is GAD65 that is the main target for GAD autoantibodies in both SPS and DM.[17] In SPS, GAD autoantibodies recognise both linear and conformational epitopes, whereas in type 1 DM they recognise only conformational epitopes.[18] An epitope is the specific region of an antigen recognised by the immune system. A linear epitope has a continuous sequence of amino acids and it is this primary structure that the autoantibody recognises. In contrast, a conformational epitope has a three-dimensional shape with discontinuous sequence of amino acids and interacts with autoantibodies based on this tertiary structure.
The GAD autoantibodies in type 1 DM recognise the carboxy-terminal end or the centre of the GAD molecule, while in SPS, the GAD autoantibodies mostly recognise the amino-terminal fragment of GAD.[17] The differential recognition of the GAD molecule is reflected in T cell immunoreactivity to it: T cells from type 1 DM patients recognise epitopes of GAD at the carboxy-terminal while those from SPS recognise epitopes of GAD at the amino-terminal.[17] GAD autoantibodies in type 1 DM tend to be restricted to the immunoglobulin G1 (IgG1) isotype, but in SPS the isotype profile is much broader, including IgG4 and IgM; these differences may either be disease-related or may reflect the much higher GAD titre in SPS.[17] GAD autoantibody titres in SPS are up to 50 times elevated but only 10 times in DM,[1] although not all SPS patients have high GAD autoantibody titres.[17] Of note, the GAD autoantibody titre in serum or cerebrospinal fluid (CSF) does not correlate with symptom severity,[1] and so titre monitoring is unnecessary.
GAD autoantibodies are not specific to SPS and DM, and occur also in cerebellar ataxia, myoclonus, epilepsy and several other neurological disorders (see Differential Diagnosis section). This spectrum of clinical entities results from differences in epitope recognition.[1]
Other Associated Autoantibodies
Table 1. Other autoantibodies associated with stiff person syndrome (SPS)10–12
| Gephyrin | Present in a very small proportion of the paraneoplastic variant. It is a postsynaptic cellular protein responsible for clustering of two inhibitory neurotransmitters: (1) glycine receptors in the spinal cord and (2) GABA-A receptors in the brain. |
| Amphiphysin | Present in 5% of the paraneoplastic variant. It is a synaptic vesicle protein responsible for endocytosis of the vesicle membrane after exocytosis of GABA from the axonal terminals. |
| GABA (A) receptor-associated protein | Present in 70% of SPS patients. It is a postsynaptic protein at GABA-ergic synapses, responsible for the stability and surface expression of GABA-A receptor and organises the clustering of GABA (A)-receptor by linking them to gephyrin. |
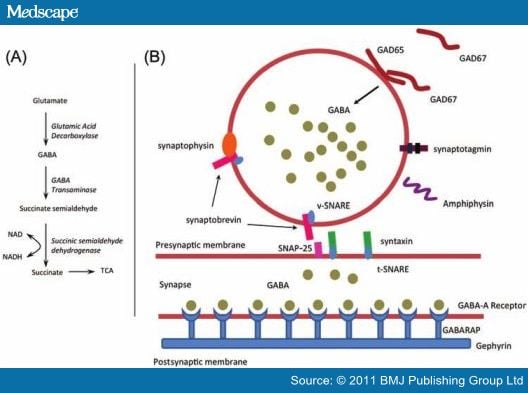
Figure 2.
A summary diagram of the γ-aminobutyric acid (GABA) metabolic pathway and the molecular biology of GABA neurotransmission.[5 10–12 19] (A) GABA biochemistry: glutamate is converted to GABA via two isoforms of glutamic acid decarboxylase (GAD65 and GAD67). GABA is then packaged into presynaptic vesicles to allow exocytosis into the GABA synapse. Excess GABA is metabolised to succinate, which then enters the tricarboxylic acid cycle. (B) The GABA synapse: GABA accumulates in the presynaptic vesicle. Fusion of vesicles with the presynaptic membrane enables exocytosis of GABA into the synapse. A protein complex is formed to allow fusion and is composed of synaptosomal-associated protein 25 (SNAP-25), synaptobrevin and syntaxin. The component proteins in this complex are anchored by SNAP receptor (SNARE) proteins; v-SNARE is vesicle-associated and t-SNARE is 'target' or presynaptic membrane-associated. Synaptotagmin is involved in docking of the vesicle with the presynaptic membrane and calcium-mediated fusion. Synaptophysin is a ubiquitous synaptic vesicle glycoprotein. Amphiphysin is involved in endocytosis and cycling of synaptic vesicle membrane, following GABA exocytosis. Once GABA has entered the synapse, it binds to GABA receptors. Gephyrin is thought to be involved with clustering of GABA receptors at the postsynaptic membrane, as is the GABA (A) receptor-associated protein (GABARAP). Autoantibodies against GAD, amphiphysin, gephyrin and GABARAP have all been identified in patients diagnosed with stiff person syndrome.
Clinical Features
The symptom onset is typically insidious. Patients may report intermittent aching and tightness in the neck, paraspinal and abdominal muscles. The rigidity spreads slowly through the proximal muscles and is often asymmetrical.[1] Over time, activities of daily living become severely impaired and patients report difficulty dressing, walking and bending forward, for example, when putting on shoes.
The age of onset varies considerably but is usually in adulthood, at an average age of 41.2 years (range 29–59 years).[20] SPS affects twice as many women as it does men,[20] in line with other adult-onset autoimmune diseases: this observation led to a nomenclature change from stiff man syndrome to SPS. Several clinical features given below are consistent mainly with GAD autoantibody positive cases.
Rigidity and Stiffness
Rigidity and stiffness of the trunk muscles are the earliest symptoms and result from constant contraction of both lumbar and abdominal muscles (figure 3). The abdominal and lumbar paraspinal rigidity begins insidiously and fluctuates at first. As it progresses, patients develop a fixed posture, giving the characteristic lumbar spine 'hyperlordosis', a diagnostic hallmark (figure 3). Importantly, lumbar hyperlordosis persists even when the patient lies flat on their back, but usually alleviates in sleep.[21]
The rigidity progresses slowly from the trunk to the proximal lower limb muscles and can cause walking difficulty. The gait may be bizarre, but typically is slow and wide, in an effort to improve balance.[1] Patients with severe and untreated SPS ultimately become bedridden through progression of the stiffness.
Facial muscle involvement is rare, but can give an 'emotionless mask' appearance.[13] If the stiffness affects the thoracic muscles, there may be restriction of chest expansion and breathing difficulty;[13] this can be catastrophic if treatment is withdrawn abruptly. The hands or feet may be involved in up to 25% of cases;[22] typically, these cases are GAD autoantibody-negative. The arms, if involved, may have a flexed posture.[3]
Superimposed Spasms
Muscle spasms, superimposed on muscle rigidity, are initially intermittent and precipitated by startle (particularly sudden auditory or tactile stimulation), by psychological factors and by passive or active movement of the affected or unaffected muscles. Spasms can be extremely painful and disabling. They are usually short lasting (minutes) and disappear gradually on removal of the triggering stimulus. Spasms may also occur in bouts, resembling tetanus,[21] or may lead to a 'shock-like' clinical presentation, with sweating, tachycardia and restlessness.[3] Falls from severe spasms are very common, sometimes even with long bone fractures and joint dislocations.[3] The fear of falling may prompt patients to use mobility aids (canes or wheelchairs), even when the severity of rigidity does not make this necessary.
Psychological Features
Clinicians treating patients with SPS must be aware of frequent coexisting psychiatric symptoms such as task-specific phobias, depression and generalised anxiety disorders. Major psychological features can dominate the clinical picture and lead to the misdiagnosis of a psychogenic movement disorder.[13] However, SPS patients do not typically have premorbid phobias or anxiety and often have realistic and appropriate fears of certain situations because of their stiffness, spasms and falls.[23] Anxiety-provoking examples include crossing roads, initiating walking without support or descending stairs without a banister.[20] The anxiety encountered in SPS therefore results more from the primary neurological disorder than from a primary phobic disorder, although the reduction in GABA levels may predispose then to an exaggerated anxiety response.[23] These associated features can complicate the diagnosis and lead to suspicions of malingering in undiagnosed cases, often reinforced by the response to diazepam and the pain relief with morphine. As with other chronic disorders, depression commonly coexists in SPS patients and should be sought and treated.[24] The psychological/psychiatric symptoms may be managed in partnership with a psychiatrist once the SPS diagnosis is secure.
Pain
Pain is common in SPS. The first symptom may be a persistent, progressively worsening ache, localised to the area of rigidity. The pain is typically chronic but worsens acutely with muscle spasms. The clinician must therefore specifically ask about pain since it significantly impacts on quality of life.
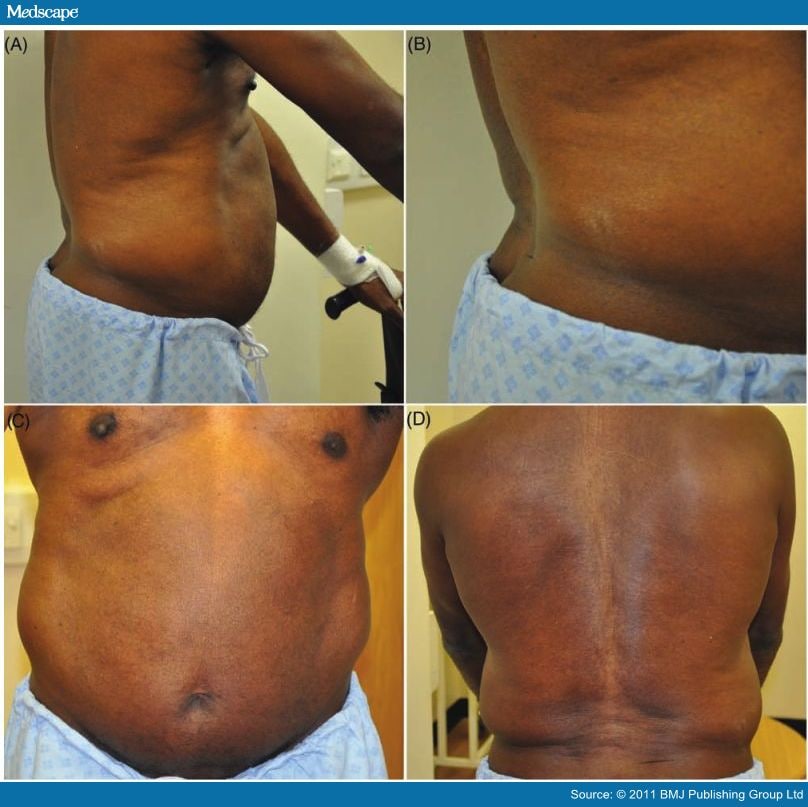
Figure 3.
Postural abnormalities and examination findings in stiff person syndrome. (A) and (B) Hyperlordosis. (C) Coexistent contraction of abdominal muscles. (D) Note the skin creases in the lumber region of the back, hinting at exaggerated lordosis.
Examination Findings
The head retraction reflex occurs in many SPS patients (see Box 1).[25] This is a non-specific abnormal cutaneo-muscular brainstem reflex, elicited by tapping the nasal ridge, upper lip, glabella or chin, provoking a backward jerk of the head or truncal retropulsion.
Eye movement disturbances may occur in SPS but are not sufficiently frequent to be a diagnostic guide. These present as gaze palsies suggestive of third, fourth or sixth nerve palsies, supranuclear gaze palsies or nystagmus.[26] An eye movement abnormality should prompt consideration of one of the 'SPS plus' variants or additional pathology (figure 4).
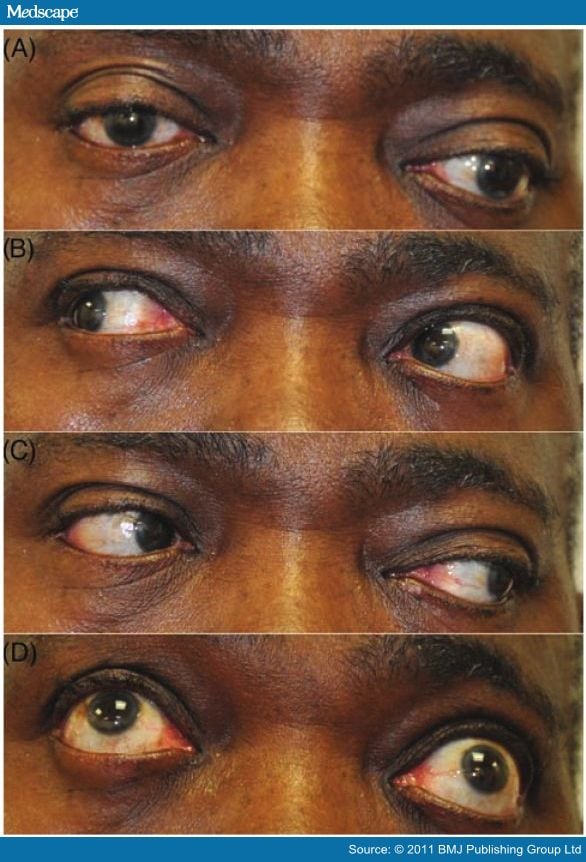
Figure 4.
Possible additional ophthalmic signs. (A) Primary position: left eye deviated down and out. (B) and (C) Rightward and leftward gaze: left eye cannot fully adduct. (D) Upward gaze: left eye fails to fully elevate. Cranial nerve palsies can be observed very occasionally in stiff person syndrome (SPS) and are more common in the SPS plus variant, progressive encephalitis with rigidity (see Differential Diagnosis section). However, this patient has Graves' ophthalmopathy with coexistent autoimmune thyroid disease.
Social Implications
Up to 65% of SPS patients cannot independently undertake normal activities of daily living,[1] due to disabling rigidity and stiffness, task-specific or non-specific phobias, the unpredictability of dangerous muscle spasms, or frequent falls.[1] Even SPS patients receiving treatment can have markedly reduced quality of life, owing to their problems in social and physical functioning.[24] Stiffness severity can have a major effect on quality of life: for example, severe cervical rigidity may limit head rotation, and cause problems when driving.[4] Coexisting depression can further diminish quality of life.
Diagnosis and Diagnostic Criteria
SPS is largely a clinical diagnosis, facilitated by a high degree of suspicion; there are no specific neurological signs or laboratory tests.[20] The variability in clinical presentation and recognition of SPS variants further increase the diagnostic uncertainty.[27] The delay in diagnosis ranges from 1 to 18 years, with a mean of 6.2 years.[20]
The Dalakas criteria[1 20] (see Box 2) are used worldwide to diagnose SPS. Patients not meeting these criteria (eg, without the classical axial distribution of stiffness and rigidity) are described as atypical.
Box 2. The Dalakas criteria for the diagnosis of typical stiff person syndrome1
|
* GAD, glutamic acid decarboxylase.
Autoimmune and Other Associations
SPS is strongly associated with other autoimmune diseases: about 35% of SPS patients also have type 1 DM.[1] Importantly, most patients with adult-onset type 1 DM do not require treatment with insulin, whether they have SPS or not.[28]About 5–10% of patients also have autoimmune thyroid disease, Graves' disease, pernicious anaemia or vitiligo.[16] Ten per cent of GAD autoantibody-positive SPS patient have epilepsy, possibly from functional impairment of GABA-ergic neurons,[16] and a further 10% have ataxia.[1]
Investigations
Details of routine and additional investigations are listed in Table 2.
Table 2.
| Tests | Essential | Occasionally needed |
|---|---|---|
| Blood tests | Full blood count, electrolytes, liver function test, thyroid function tests, fasting glucose, creatine kinase, erythrocyte sedimentation rate, C reactive protein | Protein electrophoresis, serum B12, folate, HTLV-1, treponemal agglutination, rheumatoid factor |
| Oral glucose tolerance test | If the patient does not have DM, a glucose tolerance test, together with a C-peptide curve, can usefully assess pancreatic β-cell reserve in incipient DM. | |
| Serology | Anti-GAD | In GAD-negative patients, antigephyrin and anti-amphiphysin (part of the screen for paraneoplastic SPS). Anti-GABARAP and anti-Ri might be considered in patient with atypical presentations |
| Antiparietal cell, anti-tissue transglutaminase, anti-intrinsic factor, antithyroid microsomal autoantibodies, antinuclear, extractable nucleic antigens (to identify coexisting autoimmune disease) | ||
| Cerebrospinal fluid | Cell count (normal), protein (normal), glucose (normal), locally synthesised oligoclonal IgG bands (positive) | |
| Imaging | MRI scans of brain and spinal cord (pathology here can present with muscle rigidity) | Chest x-ray, spinal x-ray, CT scan of chest/abdomen/pelvis, colonoscopy/gastroscopy, mammography/breast ultrasound scan, thyroid ultrasound scan/nuclear imaging, lymph node biopsy, whole body FDG-PET scan (to identify a possible primary tumour) |
| Electromyography | Continuous muscle fibre activity (pathognomic of SPS) | |
| Genetics | HLA-DRB1 and HLA-DQB1 genes (associated with SPS). Differential diagnosis includes hereditary spastic paraparesis (eg, SPG11) and primary dystonia (DYT1), | |
| Histopathology | Muscle biopsy (usually normal) | |
| Postmortem: often normal: inconsistent and non-specific findings such as neuronal loss in the spinal cord. |
Differential Diagnosis
The differential diagnosis of SPS is summarised in Box 3. Patients are often initially suspected of having a more common neurological, medical or psychiatric disorder. They may be referred to numerous specialists before SPS is diagnosed, for example to orthopaedic surgeons or rheumatologists for back pain and spinal stiffness, or psychiatrists for phobias and anxiety.
Box 3. Differential diagnosis
- Clinical differential
- Myelopathy: compressive, ischaemic, haemorrhagic and inflammatory (including multiple sclerosis and infectious causes)
- Myopathy: channelopathies, inflammatory, myotonic dystrophy, paramyotonia
- Neuropathic: neuromyotonia, Isaac's syndrome
- Parkinson's disease or Parkinson-plus syndromes (eg, progressive supranuclear palsy, multiple system atrophy)
- Primary lateral sclerosis
- Dystonia (generalised and focal)
- Ankylosing spondylitis
- Neuroleptic malignant syndrome, malignant hyperthermia and serotonin syndrome
- Tetanus
- Psychogenic
- Hereditary spastic paraparesis
- Leukodystrophies
- Drug-induced and toxicity: monoamine oxidase inhibitors, phenothiazines, amphetamines, 5,6-methylenedioxy-N-methyl-2-aminoindane, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine, carbon monoxide
- Spinal interneuronitis with rigidity
- Diseases associated with positive GAD autoantibodies
- Cerebellar ataxia
- Epilepsy
- Limbic encephalitis
- Myasthenia gravis
- Myoclonus
- Neuromyotonia
- Batten's disease
- GAD, glutamic acid decarboxylase.
SPS Variants
Barker et al divided SPS into three subcategories: the SPS, the stiff limb syndrome and progressive encephalomyelitis with rigidity.[27]
Stiff Limb Syndrome
There is a focal onset, usually in one lower limb, with subsequently more widespread symptoms. The stiffness usually remains most prominent in the limb where symptoms began.[1 27] Up to half of these patients have sphincter disturbance and about a third develop brainstem involvement.[27] Electrophysiological findings are similar to those in classical SPS. Most patients are GAD autoantibody-negative and only partially respond to GABA-ergic treatment.[27]Progressive Encephalitis With Rigidity
Progressive encephalomyelitis with rigidity can present in patients already suspected of having SPS. It presents as a complex progressive illness dominated by an acute onset of painful rigidity and muscle spasms in the limbs and trunk. It has a rapid course,[29] with brainstem dysfunction (nystagmus, opsoclonus, ophthalmoparesis, deafness, dysarthria, dysphagia) and profound autonomic disturbance.[30]The CSF shows a mild lymphocytic pleocytosis with elevated protein and oligoclonal IgG bands. MRI may show increased signal intensity throughout the spinal cord and the brainstem.[31] The response to diazepam is mild compared with that in classical SPS. However, the symptoms improve dramatically with methylprednisolone, suggesting that the underlying mechanism is autoimmune mediated inflammation.[30 31]
Paraneoplastic Variants
Paraneoplastic SPS, comprising 5% of patients, manifests as stiffness mostly in the neck and arms, in contrast to the distribution of typical SPS.[1] Paraneoplastic SPS is associated with malignancies of the breast, colon, lung, thymus and in Hodgkin's lymphoma, occasionally manifesting before the cancer does. Autoantibodies against amphiphysin and gephyrin may occur: this situation should prompt thorough investigation (see Table 2) and ongoing vigilance.Less common SPS variants include: persistent focal stiff man or stiff leg syndrome, a cerebellar subtype with truncal ataxia, gait ataxia, dysarthria and abnormal eye movements, and the 'jerking stiff man syndrome'.
Treatment of SPS
SPS treatments are aimed at symptom relief and/or modulation of the underlying aberrant immune process. The rarity of SPS makes it difficult to recruit sufficient patient numbers for good quality clinical drug trials, hence limiting the quality of treatment guidance. The last 30 years have provided some insight into the various options available and are the foundation upon which current practice is based (see Table 3). There remains an important role for a multidisciplinary approach to management, including physiotherapy and occupational therapy input.
Improving Symptoms: Muscle Relaxants and Other Agents
Benzodiazepines Benzodiazepines augment GABA-dependent pathways and are both anticonvulsant and anxiolytic. They are also profound muscle relaxants and have long been a mainstay treatment of SPS. Drugs such as diazepam remain preferred agents for symptom management in SPS. Over time, patients often need increasing doses for symptom relief, sometimes with troublesome side effects. Some patients require and can tolerate very large doses, but uptitration must be undertaken gradually. In general, the trend is for addition or substitution with other therapeutic agents to avoid unwanted effects.Baclofen Baclofen is a GABA-B agonist, frequently used to treat spasticity, along with benzodiazepines. Twenty years ago, it was proposed as a treatment for SPS. Most patients are maintained on oral baclofen. Sometimes high doses are needed, which may cause disabling cognitive side effects. Due to poor CSF bioavailability, intrathecal baclofen is used for severe spasticity and can significantly improve the features of both classical SPS and its variant, progressive encephalitis with rigidity and myoclonus.[32]
Silbert et al undertook a double-blind placebo-controlled trial of intrathecal baclofen in three patients with SPS. While only one patient reported subjective improvement, all three patients showed significant electrophysiological evidence of improvement and a trend towards improved muscle stiffness on objective scales.[33]
Intrathecal baclofen is typically prescribed to patients requiring high-dose benzodiazepines but experiencing intolerable side effects. Clinicians must be cautious when using intrathecal baclofen since interruption in drug delivery can lead to severe symptomatic withdrawal state and even death from autonomic failure.[34] Catheter malfunctions occurs in up to 40% of patients requiring intrathecal infusion devices for spasticity; there is a useful protocol for systematic checks if this is suspected.[35]
Other Options Dantrolene and tizanidine have been traditionally used to manage conditions with spasticity, including SPS. These are commonly combined with other muscle relaxants. Tiagabine, gabapentin, valproate and carbamazepine may all help SPS symptoms; vigabatrin is rarely used because of its potential for visual field constriction. Levetiracetam has been the subject of a single-blind placebo-controlled trial in three patients and benefited both symptoms and electrophysiological findings.[36] Propofol helped the stiff limb variant of SPS in a single case refractory to other therapeutic strategies. Intramuscular botulinum toxin A can significantly improve muscle tone and spasms, ambulation and pain in SPS. Analgesics remain an important part of SPS treatment. However, clinicians should be aware that opiate analgesics, while reducing the pain of rigidity and spasms may, on rare occasions, worsen these symptoms; follow-up monitoring is advisable when commencing or uptitrating these drugs.
Disease Modifying Immunomodulation/Immunosuppression
Intravenous Immunoglobulin Intravenous immunoglobulin (IVIG) is the best second-line treatment for patients with severe or refractory SPS. The evidence for this came originally from several case reports where there were significant improvements in stiffness, startle, functional status and clinical examination findings, as well as most showing radiographic and serological improvements. IVIG has subsequently been shown to improve quality of life in SPS and also to improve symptoms in the GAD-positive stiff limb variant.A randomised, double-blinded, placebo-controlled, crossover trial of monthly IVIG demonstrated a significant decrease in stiffness, which stabilised during washout and increased again on switching to placebo. IVIG-treated patients reported improvement of symptoms and ability to undertake activities of daily living, lasting between 6 weeks and 1 year. The GAD autoantibody titre also fell after IVIG.[37]
While IVIG is generally considered safe, neurologists should be aware of its common and important adverse effects. These include immediate infusion reactions (mild to severe) with a small but potentially risk of fatal anaphylaxis. This occurs typically in IgA-deficient patients, which is therefore a relative contraindication for IVIG. There may also be skin reactions, headaches, aseptic meningitis and renal tubular acidosis. Venous thromboembolic disease is a significant risk, particularly in those with limited mobility. Arterial thrombus formation may lead to stroke, myocardial infarction, pulmonary embolism or ischaemia affecting other tissue beds.
Cost remains a major factor: treatment decisions in the UK are made on a case-by-case basis. However, the rarity of SPS means that the overall cost of treating this patient group is much less than for a more common disease such as chronic inflammatory demyelinating polyradiculoneuropathy. There have been regular updates in the published guidance on the use of IVIG in immune-mediated neuromuscular diseases in recent years. The European Federation of Neurological Sciences recommends IVIG for patients with SPS who respond incompletely to diazepam and/or baclofen and who have a significant disability requiring a cane or walker due to truncal stiffness and frequent falls.[38] The recommended dose is 2 g per kg over 2–5 days.
Plasma Exchange The evidence supporting plasma exchange for SPS is less well established than for IVIG and there have been several conflicting results. Plasma exchange was first used successfully over 20 years ago. In anecdotal reports, some patients enjoyed improvements in symptoms and serological and electrophysiological markers; however, equal numbers had no benefit. Patients showing improvement tended to be on beneficial concomitant medications. To date, there has been no reported randomised placebo-controlled study of plasma exchange in SPS.
Rituximab As with many autoimmune disorders, the disease course should, in theory, be altered by depletion of mature B cells using rituximab, the chimeric anti-CD20 monoclonal autoantibody. Rituximab was recently shown to give symptomatic and serological remission in patients with otherwise refractory SPS.[39]
Other Immunomodulatory Agents Corticosteroids have often been used in patients with SPS either as monotherapy or combined with other therapeutic agents with improvement of spasms and autoantibody titre. However, there has never been a good quality clinical trial to determine their overall role in SPS. Other immune system modulating agents give variable benefit, including mycophenolate mofetil, azathioprine, cyclophosphamide, cyclosporine, tacrolimus and sirolimus.
Table 3. Main treatment options in stiff person syndrome
| Agent | Action | Daily doses | Side effects |
|---|---|---|---|
| Drugs treating symptoms | |||
| Benzodiazepines, eg, diazepam, clonazepam | GABA-A agonist | Diazepam 5–100 mg (divided doses)Clonazepam 1–6 mg (divided doses) (though often doses are far higher) | Drowsiness, vertigo, dysarthria, respiratory depression |
| Baclofen | GABA-B agonist | Oral 5–60 mg (divided doses)Intrathecal 50–800 μg/day | Drowsiness, vertigo |
| Antiepileptics, eg, levetiracetam, gabapentin | GABA-ergic and other actions | Levetiracetam 2000 mgGabapentin 3600 mg | Variable |
| Other options: tizanidine, dantrolene, botulinum toxin | |||
| Drugs that modulate the immune process | |||
| IVIG | Incompletely understood | 2 g/kg | Infusion reactions including anaphylaxis, thrombotic events, headaches, aseptic meningitis |
| Plasma exchange | Incompletely understood | 5–6 Exchanges | Hypotension, bleeding, allergic reaction, severe immune suppression |
| Rituximab | B cell depletion | 2 g (Divided doses) | Breathing problems, arrhythmia and rarely skin reactions (Stevens–Johnson syndrome) and progressive multifocal leucoencephalopathy |
Other treatments: analgesics, physiotherapy, occupational therapy.
GABA, γ-aminobutyric acid; IVIG, intravenous immunoglobulin.
Prognosis and Future Prospects
SPS follows a variable course over 6–28 years, measured from symptom onset to either the last follow-up visit or death. Its rate of progression depends on several factors, including: (1) whether the initial presentation and symptoms are of classical SPS, (2) belong to the 'stiff man plus' category or (3) whether it is associated with disorders such as DM or malignancy.[21] Patients with classic SPS usually respond well to treatment and their condition stabilises over time, although paroxysmal autonomic dysfunction or sudden death occurs in 10% of SPS patients.[21] Autonomic dysfunction results from a succession of spasms or sudden withdrawal of medication.[40]
Although SPS is rare, it can cause significant morbidity and mortality; its pathogenesis relates to diseases with major public health impact, including DM and cancer. Further exploration of the link between SPS and DM could help to predict and classify DM, to alter the implications of its therapy and to prevent its associated morbidities.[29]
For SPS, the aim should be earlier recognition and treatment under the care of a neurologist. SPS is severely disabling, can substantially affect life expectancy and impair physical and mental capabilities. Disability results in a reduced quality of life and affects an individual's potential for education and earning. Therefore, a better understanding of the natural history, mechanisms of disease and the impact of disease progression over time, along with the long-term effects of treatment, are needed to make further progress.
References
- Dalakas MC. Stiff person syndrome: advances in pathogenesis and therapeutic interventions. Curr Treat Options Neurol 2009;11:102–10.
- Moersch FP, Woltman HW. Progressive fluctuating muscular rigidity and spasm ("stiff-man" syndrome); report of a case and some observations in 13 other cases. Proc Staff Meet Mayo Clin 1956;31:421–7.
- Gordon EE, Januszko DM, Kaufman L. A critical survey of stiff-man syndrome. Am J Med 1967;42:582–99.
- Lorish TR, Thorsteinsson G, Howard FM Jr.. Stiff-man syndrome updated. Mayo Clin Proc 1989;64:629–36.
- Solimena M, Folli F, Denis-Donini S, et al. Autoantibodies to glutamic acid decarboxylase in a patient with stiff-man syndrome, epilepsy, and type I diabetes mellitus. N Engl J Med 1988;318:1012–20.
- Howard FM Jr.. A new and effective drug in the treatment of the stiff-man syndrome: preliminary report. Proc Staff Meet Mayo Clin 1963;38:203–12.
- Piccolo G, Cosi V, Zandrini C, et al. Steroid-responsive and dependent stiff-man syndrome: a clinical and electrophysiological study of two cases. Ital J Neurol Sci 1988;9:559–66.
- Vicari AM, Folli F, Pozza G, et al. Plasmapheresis in the treatment of stiff-man syndrome. N Engl J Med 1989;320:1499.
- Amato AA, Cornman EW, Kissel JT. Treatment of stiff-man syndrome with intravenous immunoglobulin. Neurology 1994;44:1652–4.
- Raju R, Rakocevic G, Chen Z, et al. Autoimmunity to GABAA-receptor-associated protein in stiff-person syndrome. Brain 2006;129:3270–6.
- Folli F, Solimena M, Cofiell R, et al. Autoantibodies to a 128-kd synaptic protein in three women with the stiff-man syndrome and breast cancer. N Engl J Med 1993;328:546–51.
- Butler MH, Hayashi A, Ohkoshi N, et al. Autoimmunity to gephyrin in Stiff-Man syndrome. Neuron 2000;26:307–12.
- Levy LM, Dalakas MC, Floeter MK. The stiff-person syndrome: an autoimmune disorder affecting neurotransmission of gamma-aminobutyric acid. Ann Intern Med 1999;131:522–30.
- Thompson PD. The stiff-man syndrome and related disorders. Parkinsonism Relat Disord 2001;8:147–53.
- Nollet H, Vanderstraeten G, Sustronck B, et al. Suspected case of stiff-horse syndrome. Vet Rec 2000;146:282–4.
- Solimena M, Folli F, Aparisi R, et al. Autoantibodies to GABA-ergic neurons and pancreatic beta cells in stiff-man syndrome. N Engl J Med 1990;322:1555–60.
- Lohmann T, Hawa M, Leslie RD, et al. Immune reactivity to glutamic acid decarboxylase 65 in stiffman syndrome and type 1 diabetes mellitus. Lancet 2000;356:31–5.
- Ellis TM, Atkinson MA. The clinical significance of an autoimmune response against glutamic acid decarboxylase. Nat Med 1996;2:148–53.
- Alberts B, Johnson A, Lewis J, et al. Molecular Biology of the Cell. 4th edition. New York, NY: Garland Science Publishing Company 2001:711–26.
- Dalakas MC, Fujii M, Li M, et al. The clinical spectrum of anti-GAD antibody-positive patients with stiff-person syndrome. Neurology 2000;55:1531–5.
- Gershanik OS. Stiff-person syndrome. Parkinsonism Relat Disord 2009;15(Suppl 3):S130–4.
- Meinck HM. Stiff man syndrome. CNS Drugs 2001;15:515–26.
- Ameli R, Snow J, Rakocevic G, et al. A neuropsychological assessment of phobias in patients with stiff person syndrome. Neurology 2005;64:1961–3.
- Gerschlager W, Schrag A, Brown P. Quality of life in stiff-person syndrome. Mov Disord 2002;17:1064–7.
- Ruivard M, Berger C, Achaibi A, et al. Physician compliance with outpatient oral anticoagulant guidelines in Auvergne, France. J Gen Intern Med 2003;18:903–7.
- Economides JR, Horton JC. Eye movement abnormalities in stiff person syndrome. Neurology 2005;65:1462–4.
- Barker RA, Revesz T, Thom M, et al. Review of 23 patients affected by the stiff man syndrome: clinical subdivision into stiff trunk (man) syndrome, stiff limb syndrome, and progressive encephalomyelitis with rigidity. J Neurol Neurosurg Psychiatr 1998;65:633–40.
- Leslie RD. Predicting adult-onset autoimmune diabetes: clarity from complexity. Diabetes 2010;59:330–1.
- Brown P, Marsden CD. The stiff man and stiff man plus syndromes. J Neurol 1999;246:648–52.
- Gouider-Khouja N, Mekaouar A, Larnaout A, et al. Progressive encephalomyelitis with rigidity presenting as a stiff-person syndrome. Parkinsonism Relat Disord 2002;8:285–8.
- McCombe PA, Chalk JB, Searle JW, et al. Progressive encephalomyelitis with rigidity: a case report with magnetic resonance imaging findings. J Neurol Neurosurg Psychiatr 1989;52:1429–31.
- Stayer C, Tronnier V, Dressnandt J, et al. Intrathecal baclofen therapy for stiff-man syndrome and progressive encephalomyelopathy with rigidity and myoclonus. Neurology 1997;49:1591–7.
- Silbert PL, Matsumoto JY, McManis PG, et al. Intrathecal baclofen therapy in stiff-man syndrome: a double-blind, placebo-controlled trial. Neurology 1995;45:1893–7.
- Meinck HM, Tronnier V, Rieke K, et al. Intrathecal baclofen treatment for stiff-man syndrome: pump failure may be fatal. Neurology 1994;44:2209–10.
- Bardutzky J, Tronnier V, Schwab S, et al. Intrathecal baclofen for stiff-person syndrome: life-threatening intermittent catheter leakage. Neurology 2003;60:1976–8.
- Sechi G, Barrocu M, Piluzza MG, et al. Levetiracetam in stiff-person syndrome. J Neurol 2008;255:1721–5.
- Dalakas MC, Fujii M, Li M, et al. High-dose intravenous immune globulin for stiff-person syndrome. N Engl J Med 2001;345:1870–6.
- Elovaara I, Apostolski S, van Doorn P, et al. EFNS guidelines for the use of intravenous immunoglobulin in treatment of neurological diseases: EFNS task force on the use of intravenous immunoglobulin in treatment of neurological diseases. Eur J Neurol 2008;15:893–908.
- Baker MR, Das M, Isaacs J, et al. Treatment of stiff person syndrome with rituximab. J Neurol Neurosurg Psychiatr 2005;76:999–1001.
- Mitsumoto H, Schwartzman MJ, Estes ML, et al. Sudden death and paroxysmal autonomic dysfunction in stiff-man syndrome. J Neurol 1991;238:91–6.
Sidebar
Practice Points
- Stiff person syndrome (SPS) is underdiagnosed and often misdiagnosed.
- The most prominent clinical finding is hyperlordosis due to lumbar paraspinal and abdominal muscle cocontraction with superimposed spasms.
- Most cases have a clear autoimmune basis, characterised by autoantibodies against glutamic acid decarboxylase and γ-aminobutyric acid (A) receptor-associated protein. Rarer paraneoplastic variants occur, with antibodies against gephyrin and amphiphysin.
- In all, 30–40% of SPS patients have, or develop, diabetes mellitus.
- The diagnosis of SPS should be based on established clinical, laboratory and electromyography criteria. Cases that do not fit within these criteria should be labelled atypical or an alternative diagnosis sought.
- Psychiatric disorders are common in SPS; referral to a psychiatrist may be necessary as part of the long-term management strategy.
- Therapeutic approaches include symptomatic therapy and immunomodulatory therapy. Some patients require (and can tolerate) very large doses of diazepam; combination symptomatic treatments are often necessary. Intravenous immunoglobulin is a mainstay treatment for those refractory to symptomatic treatment and is used increasingly early in the disease.


